

05月09 百邁客亞洲棉科研成果在Nature Genetics上發表
棉花是世界上最重要的經濟作物之一,在2018年5月8日中國農業科學院棉花研究所所長李付廣研究員、武漢大學朱玉賢院士、中國農業科學院棉花研究所杜雄明研究員、中國農業科學院農業基因組研究所所長黃三文研究員、林濤博士與北京百邁客生物科技有限公司關于亞洲棉的合作成果發表在Nature Genetics上,論文題目為“Resequencing of 243 diploid cotton accessions based on an updated A genome identifies the genetic basis of key agronomic traits”。該研究以全新亞洲棉基因組為基礎,增加遺傳進化和GWAS研究,對棉的品質、產量、抗病等重要農藝性狀進行研究。其中朱玉賢院士與李付廣研究員為通訊作者,杜雄明研究員為第一作者。
以下為文獻詳細解讀:
英文題目:Sequencing of 243 diploid cotton accessions based on an updated A genome identifies the genetic basis of key agronomic traits.
中文題目:以更新的亞洲棉A基因組為基礎的243份二倍體棉花的重要農藝性狀的研究
1. 摘要:
亞洲棉(Gossypium arboreum)和草棉(Gossypium herbaceum)的祖先是現代栽培異源四倍體棉花A亞基因組的供體。本研究中通過整合了不同的技術,提升了亞洲棉的基因組組裝水平;同時對243株二倍體棉花(亞洲棉和草棉)進行全基因組重測序分析,繪制基因組變異圖譜,并發現亞洲棉和草棉(A)與雷蒙德氏棉(D)同時進行了分化;單獨的對亞洲棉分析表明亞洲棉起源于中國南部,隨后被引入長江和黃河地區,大多數具有馴化相關特性的種質都經歷了地理隔離;通過亞洲棉的全基因組關聯分析(GWAS),鑒定了亞洲棉11個重要農藝性狀的98個顯著關聯位點,GaKASIII的非同義替換(半胱氨酸/精氨酸替換)使得棉籽中的脂肪酸組成(C16:0和C16:1)發生了變化;棉花枯萎病抗性與GaGSTF9基因的表達激活相關。本研究對理解棉花A亞基因組的進化具有重要的意義。
2. 研究背景:
棉花是世界上最重要的商業作物之一,同時也是研究植物多倍化的有價值的資源。亞洲棉最可能在馬達加斯加或印度河流域文明(巴基斯坦摩亨佐達羅)開始馴化,隨后分散到非洲和亞洲一些地區。亞洲棉最初在1000多年前作為觀賞植物引入中國。當地方的農業生態環境的適應和人類選擇影響的過程中,中國的Gossypium arboreum形成了獨特的地理種群,稱之為“sinense cotton”。
雖然棉花種植者已經基于RFLP和SSR markers構建了各種遺傳圖譜,但是G. arboreum和G. herbaceum優良農藝和經濟性狀的基因尚未被鑒定。同樣通過種內和種間特異性雜交將二倍體的重要特性引入四倍體并不是富有成效的,G. raimondii,G. arboreum,G. hirsutum和G. barbadense基因組序列的發布為研究群體遺傳,栽培和馴化提供了先決條件。通過GWAS和QTLs在水稻,玉米,大豆,谷子,黃瓜,番茄和陸地棉中鑒定了許多候選基因。本研究中,利用了三代PacBio和Hi-C技術,重新組裝了高質量的亞洲棉基因組,分析了243份二倍體棉花種質的群體結構和基因組分化趨勢,同時確定了一些有助于棉花皮棉產量遺傳改良的候選基因位點。
3. 材料和方法:
測序材料:
基因組測序材料:二倍體G. arboreum栽培品種cultivar Shixiya1(SXY1);
自然群體材料選擇:243份棉花,包含230份亞洲棉 G. arboretum 和13份草棉 G. herbaceum [243份棉花選自國家種質基因庫(中國安陽),種植在中國農業科學院棉花研究所(ICR,CAAS)的溫室中],插入片段長度500 bp;測序深度6X;
遺傳群體材料選擇:親本(GA0146和GA0149),測序深度20X;2個混池(F2群體,有絨型和無絨型各20個子代),測序深度30X;
群體材料表型調查:
在230份亞洲棉中選擇了215份表型穩定的材料,分別在河南安陽,海南三亞和新疆阿克蘇進行種植,大部分性狀選自多年多點的表型數據進行調查,每個地點設置3次重復。
測序平臺:
PacBio RSII和Illumina HiSeq 2500
相關軟件:
基因組組裝(Canu和Falcon;Quiver;Pbjelly);TEs轉座元件注釋(RepeatScout,LTR-FINDER,MITE和PILER;Repbase;REPET;RepeatMasker);基因預測注釋(geMoMa;Augustus;PASA;EVidenceModeler;InterProScan)
群體研究:比對注釋(BWA,Picard,GATK,ANNOVAR);群體結構分析(FastTree,PHYLIP,STRUCTURE);連鎖不平衡分析(Haploview);遺傳多樣性分析(π,Fst);全基因組關聯分析(EMMAX);
4. 研究結果:
1. 亞洲棉基因組組裝更新
三代+Hi-C:PacBio reads ?(77.6×);有效Hi-C reads(>20×)
三代組裝結果:共計獲得了142.54 Gb ?原始三代測序數據,組裝1.71 Gb亞洲棉基因組,Contig N50=1.1 Mb,最長的Contig為12.37 Mb
(1)Hi-C輔助基因組組裝:利用Hi-C技術將組裝的1573 Mb的數據定位到13條染色體上,與已經發表的基因組相比,當Hi-C數據比對到更新的基因組后,對角線外的不一致性明顯減少(圖1 a-b)。
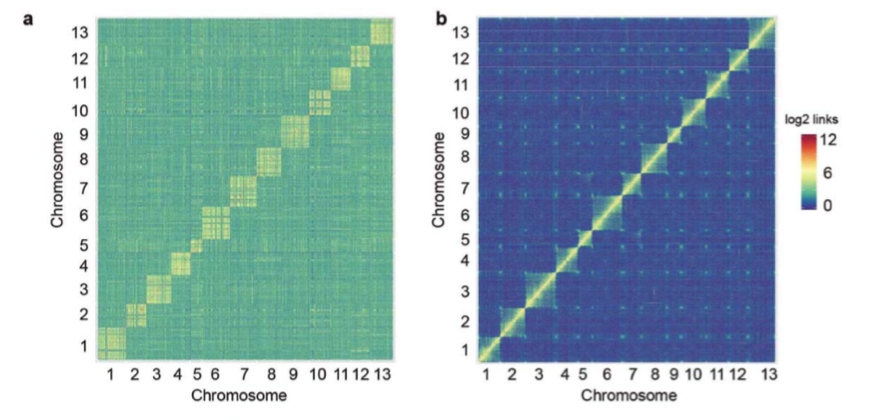
圖1 Hi-C數據在兩版亞洲棉基因組上的比對
a. Hi-C數據與亞洲棉原基因組比對;b. Hi-C數據與亞洲棉更新基因組比對
(2)基因組共線性分析:進行了亞洲棉A型與異源四倍體陸地棉的AADD型的共線性分析,發現更新后的基因組的共線性更高(圖2 a-b)。
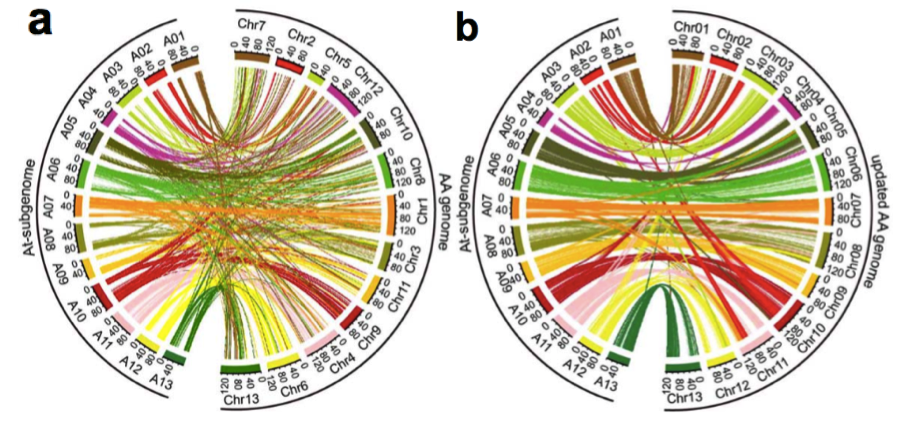
圖2 亞洲棉(AA型)與陸地棉(AADD型)共線性分析
a. 亞洲棉原基因組與陸地棉基因組共線性分析;b. 亞洲棉更新基因組與陸地棉基因組共線性分析
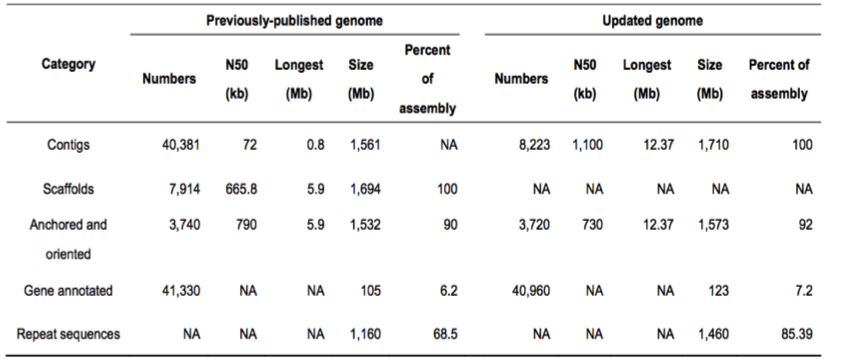
(3)亞洲棉原基因組(二代)與更新后基因組(三代)比較(表1):
表1 亞洲原基因組與更新后基因組的組裝指標比較
2. 二倍體棉花群體遺傳進化分析
(1)二倍體棉花群體材料選擇
共計選擇了243份二倍體棉花材料:230份亞洲棉G. arboreum (A2) ?和13份草棉G. herbaceum (A1);材料來源:中國南部(SC),長江(YZR)和黃河(這些區域代表了中國二倍體棉花大部分的表型和地理多樣性)。
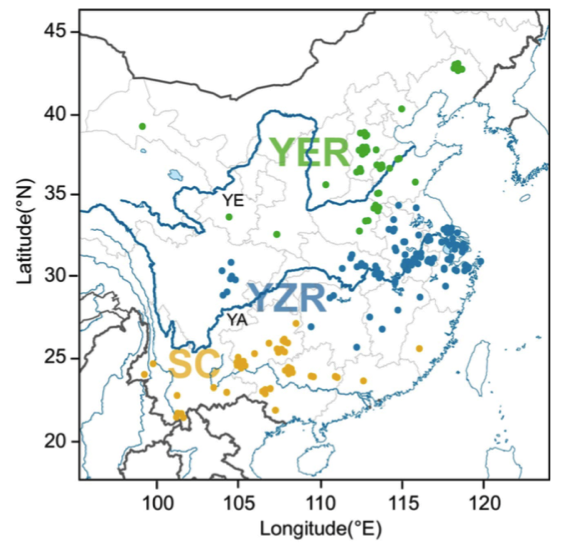
圖3 亞洲棉的地理分布
(2)群體重測序數據統計
通過重測序的研究策略,利用Illumina HiSeq 2500 ?測序平臺,PE125雙端測序,共計獲得了2.29 T數據,平均測序深度~6.0×,以本研究中更新后的亞洲棉基因組為參考基因組進行比對分析,統計獲得了17,883,108個高質量SNPs和2,470,515個indels,242,449 個SNPs(1.36%)和16,816個indels(0.68%)位于亞洲棉36,205個基因的編碼區。在31,549個基因中,共計鑒定了128,512(0.72%)個非同義突變SNPs,在8,117個基因中共計鑒定了11,372 (0.46%)個indels。
(3)二倍體棉花群體分層分析
以雷蒙德氏棉(G. raimondii)為外群,利用72,419個SNPs構建系統發育樹,顯示:G. herbaceum(草棉)和G. arboretum(亞洲棉)聚類成2個獨立的群(圖4 a-b)。G. arboretum(亞洲棉)進一步又分為SC,YZR和YER三個群,顯示了地理分布模式的差異,進而利用PCA分析支持這一結果(圖4 c)。同時發現了亞洲棉和草棉是由不同的祖先種馴化而形成。
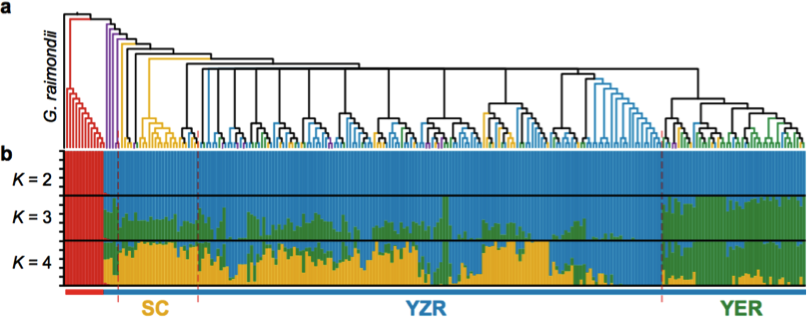
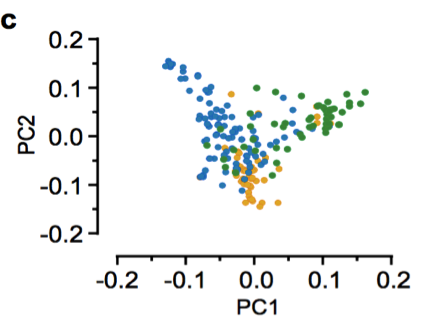
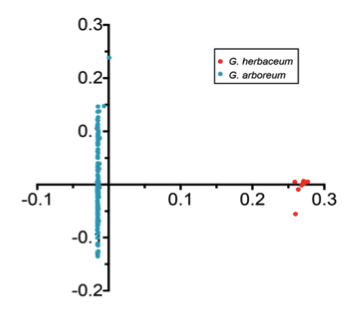
圖4 二倍體棉花的群體分層分析
a. 243份二倍體棉花系統發育樹 b. 243份二倍體棉花的群體結構分析 c. PCA主成分分析(中國亞洲棉的PCA分析;亞洲棉和草棉的PCA分析)
(4)二倍體棉花LD和選擇性清除分析
通過表型計算統計發現,與長江和黃河區域的兩個不同地區的材料項目,中國南部的材料的表型相對匱乏。此外中國南部SC的亞洲棉(π=0.211×10-3)比長江流域YZR(π=0.197×10-3)和黃河流域YER(π=0.199×10-3)的亞洲棉的核苷酸多態性高,這表明了亞洲棉最早在中國南部地區種植,并進一步擴展到長江和黃河地區,而這與之前基于SSR分析的結果一致;連鎖不平衡分析結果顯示,亞洲棉的LD衰減距離約為105.5 kb(r2=0.40),草棉的衰減距離約為145.5 kb(r2=0.39)(圖5),其LD衰減距離與大豆(約83 kb)和水稻(秈稻約123 kb;粳稻約167 kb)接近,但遠遠高于栽培玉米(22-30 kb)。大約有23.9%的亞洲棉和22.9%的草棉的等位基因與雷蒙德氏棉的基因組相一致,暗示了亞洲棉與草棉同時開始分化(圖6)。
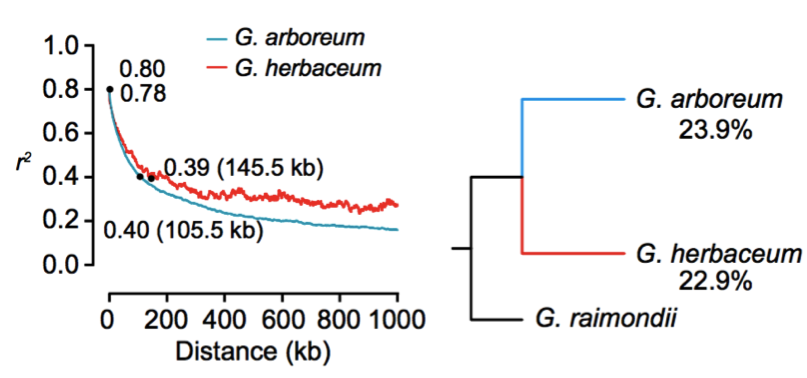
圖5 連鎖不平衡分析? ? ? ? ? ? ? ? ? ? ? ? ? ? ?圖6 棉屬的系統發育與等位基因分析
人工選擇在農作物的馴化和遷徙的過程中具有重要的作用。群體結構分析顯示當K=4時,YER與SC和YZR明顯不同(圖4 b,K=4)。通過兩兩群體間(SC vs. YZR, SC vs. YER, YZR vs. YER)的選擇性清除分析(FST)鑒定出了分別覆蓋到3,162,2,879和3,308個基因上的59,53和51個顯著遺傳分化的區域。SC和YZR之間的21個分化的區域(約43.5 Mb 含有915個基因)在群體SC和YER之間是保守的(圖7 a)。
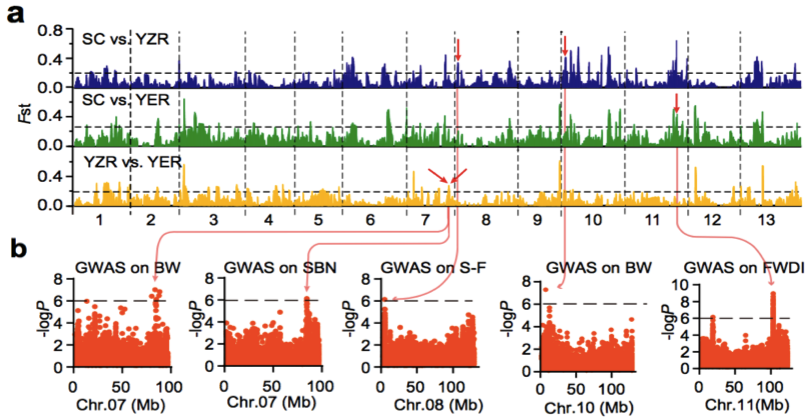
圖7 a. 亞洲棉SC,YZR和YER的選擇性清除分析;b. 全基因組關聯分析
3. 亞洲棉的全基因組關聯分析(GWAS)
對來自不同環境下的11個重要性狀進行全基因組關聯分析,在98個顯著關聯的信號中,其中25信號個來自基因區(外顯子或內含子區),包含與形態性狀相關的8個信號區,與產量性狀相關的6個信號區,與油籽性狀相關的3個信號區;剩余73個信號來自非編碼區。大部分農藝性狀的GWAS關聯信號中顯示地理差異,如交配分支數,開花期,鈴重和抗病性這些性狀定位在保守的基因區(圖7 b,表2)。因此推斷成熟度,產量和抗病性這些性狀一直處于強烈的認為和/或地理選擇之下。
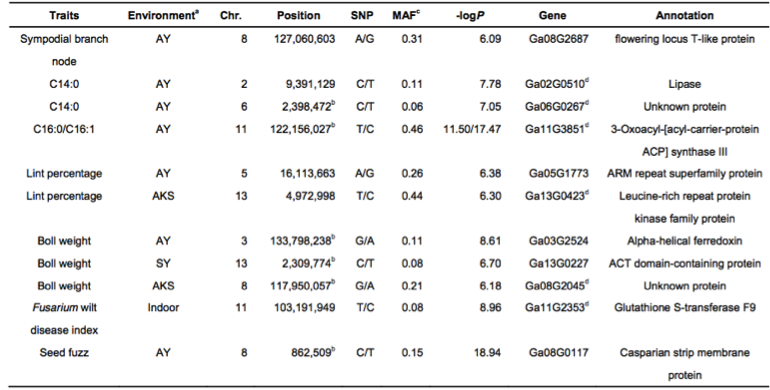
表2 部分與性狀關聯的SNPs及候選基因
(1)脂肪酸含量相關基因的定位與研究 棉花是世界上第六大植物油來源,通過GWAS關聯分析,在11號染色體上的GaKASIII基因組座位上(Ga11G3851)的第8個外顯子區獲得了1個顯著的SNP位點,該基因編碼3-Oxoacyl-[acyl-carrier-protein ACP] synthase III(3-氧酰基-[酰載體蛋白]合酶III),如圖8 a-c,KASIII基因編碼的這一關鍵酶可以使得脂肪酸鏈從C2到C4延伸,并最終確定種子中棕櫚酸(C16:0)和棕櫚油酸(C16:1)的組成。GaKASIII基因的多態性導致了保守的ACP_synthase_III_(酰載體蛋白合酶III)結構域中半胱氨酸/精氨酸間的置換(圖8 c),單倍型B(TGT,Cys)主要出現在低含油量種質中,而在高含油量種質中發現單倍型A(CGT,Arg)(圖8 d-e)。GaKASIII基因在開花后(DPA)的30天表達量最高(圖9),這是種子油量積累的關鍵階段,在單倍型種質A中,C16:0和C16:1含量以顯著的速率累積(圖10);蛋白質結構模型預測顯示,半胱氨酸/精氨酸殘基位于α螺旋處,該位點靠近酶活性位點,同時是輔酶A(CoA)結合位點(圖11)。
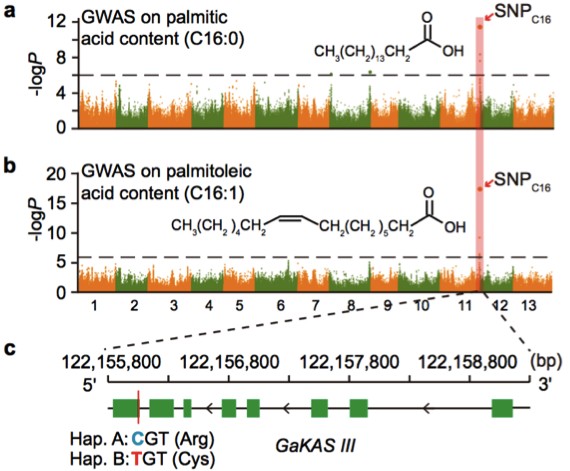
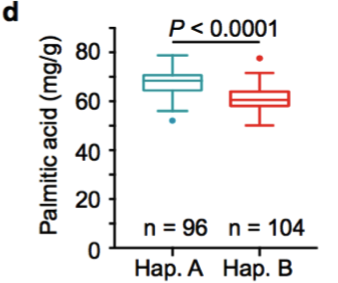
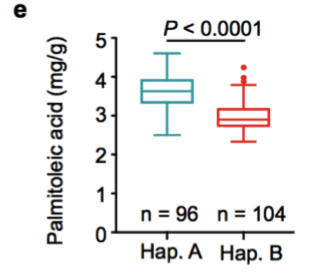
圖8 脂肪酸含量GWAS關聯分析
a. 棕櫚酸含量的GWAS關聯分析;b. 棕櫚油酸的GWAS關聯分析;c. GaKASIII基因的變異(Arg/Cys);d-e. Hap. A和Hap. B中棕櫚酸和棕櫚油酸含量比較
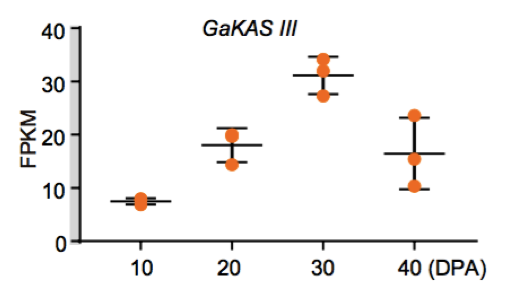
圖9 GaKASIII基因在棉花胚珠發育過程中的表達
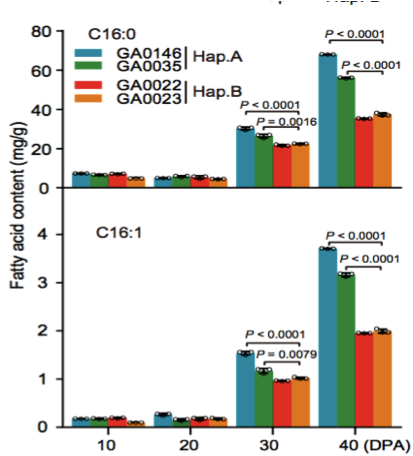
圖10 在棉花生長發育過程中C16:0和C16:1脂肪酸含量分析(Hap. A和Hap. B)
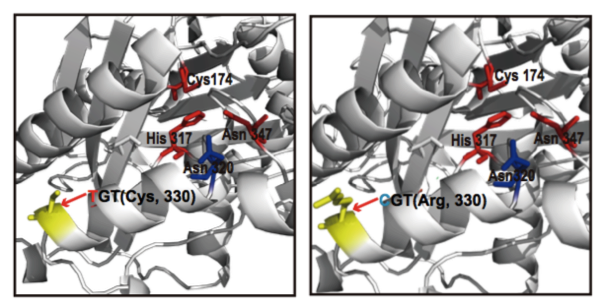
圖11 GaKASIII蛋白結構模型
(2)棉花枯萎病抗性相關基因的研究 棉花枯萎病是由尖孢鐮刀菌萎蔫專化型Fusarium oxysporum f. sp. vasinfectum (FOV)引起的棉花維管束病害,是棉花產量的重要威脅之一。通過GWAS,進行棉花FOV抗性分析,發現在11號染色體上獲得了強的關聯信號,其-logP value=8.96(圖12 a)。進一步分析表明,關聯到的SNP簇位于Ga11G2353基因的上游(圖12 b),該基因與擬南芥GSTF9基因為直系同源基因,GSTF9基因編碼參與植物對生物和非生物脅迫響應的谷胱甘肽S轉移酶(glutathione-S-transferases),攜帶疾病易感等位基因‘T’(以紫色顯示)的種質主要在SC群體中發現,所有YER群體材料攜帶耐病等位基因‘C’(以橙色顯示)(圖12 c)。研究發現GSTF9基因僅在FOV接種到亞洲棉幼苗的耐受系中上調(圖13),與空載體棉花系(TRV::00)相比,GSTF9基因沉默棉花品系(TRV::GSTF9, the virus-induced gene silencing,VIGS,vector carrying the GSTF9 gene)對于FOV的接種更加敏感(圖14-15)。此外,TRV::GSTF9植株系與TRV::00植株系相比,TRV :: GSTF9植株系中的真菌DNA的量顯著高于TRV::00植株系,且GST催化活性顯著低于TRV::00植株系(圖16 a-b),表明GaGSTF9基因可能是亞洲棉FOV抗性的靶標。
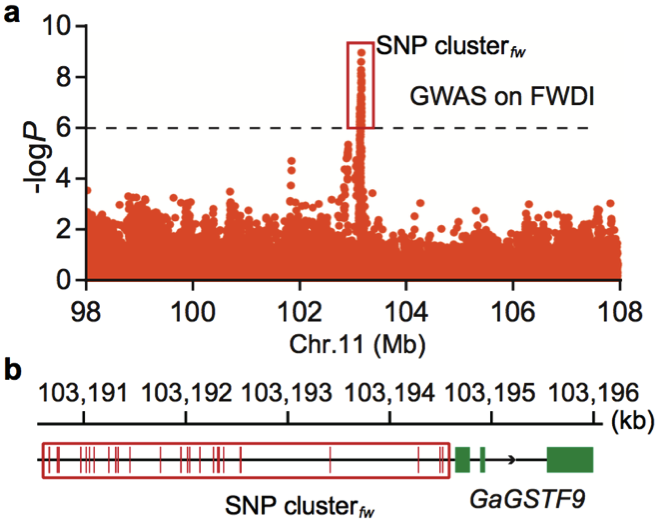
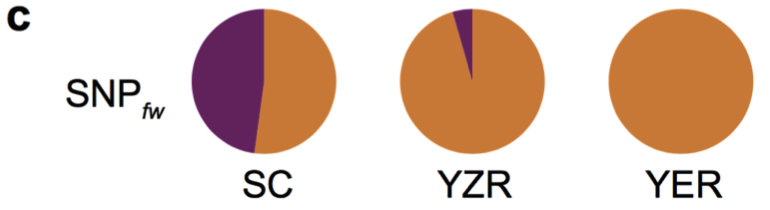
圖12 亞洲棉FWDI的全基因組關聯分析
a. FWDI的GWAS分析;b. GaGSTF9基因結構及附近關聯到的SNPs;c. 關聯分析群體基因分型 敏感型(紫);耐受型(橙)
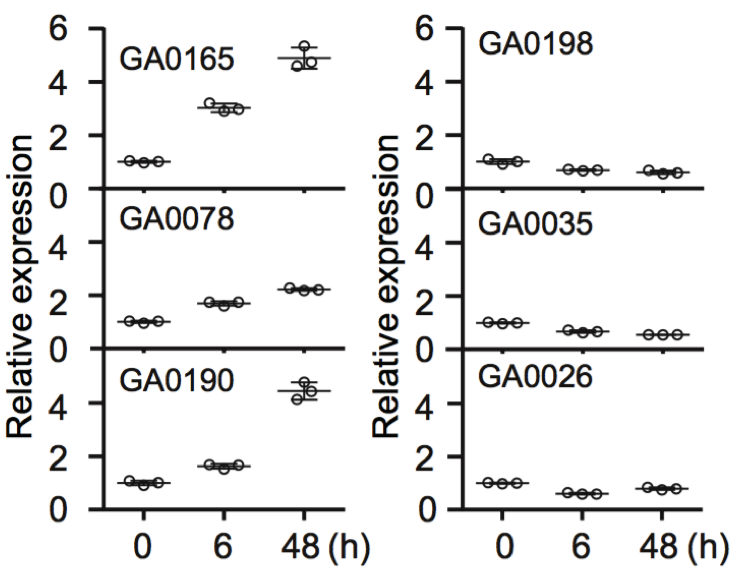
圖13 GaGSTF9基因表達的qRT-PCR分析
注:根部接種鐮刀霉菌,(高耐受型GA0165,GA0078和GA0190;高敏感型GA0198,GA0035和GA0026)
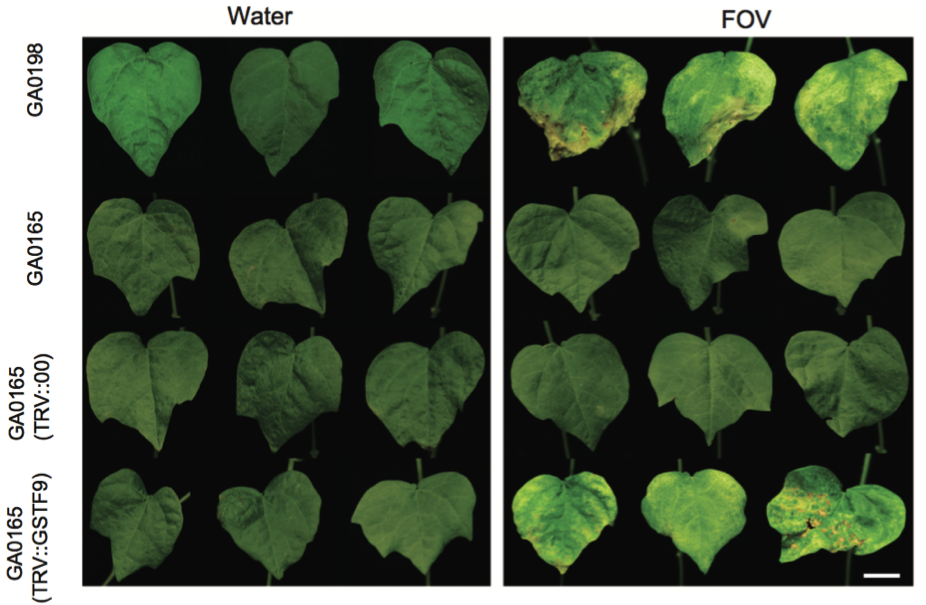
圖14 不同植株處理后的病癥比較(GA00198,GA0165,TRV::00和TRV::GSTF9,
處理條件:接種水和FOV)
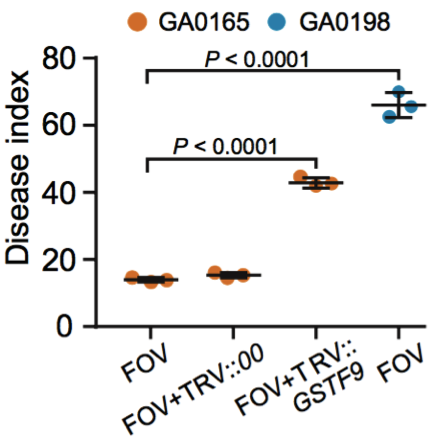
圖15 不同植株處理后的疾病感染指數比較(GA00198,GA0165,TRV::00和TRV::GSTF9,處理條件:接種FOV)
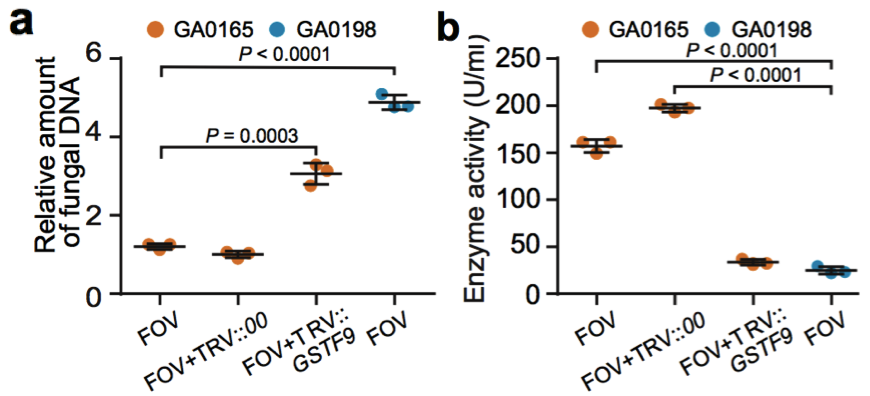
圖16 a GA00198, GA0165, TRV::00和TRV:GSTF9植株中FOV DNA的相對含量測定;b GA00198, GA0165, TRV::00和TRV:GSTF9植株中GST酶活性測定;
(3)與棉絨相關基因的研究 棉絨是覆蓋種子表面的短纖維。研究中選擇了亞洲棉種質中的158份有絨毛和57份無絨毛材料進行GWAS關聯分析,最終在8號染色體上(?0.6 Mb至?1.3 Mb的區間內)獲得了強烈的關聯信號(圖17 a-b)。QTL分析也同樣定位到8號染色體上(圖17 c)。通過有絨毛品系(GA0146)和無絨毛品系(GA0149)雜交獲得的F2代顯示了有絨毛和無絨毛的表型分離比為1:3(圖17 d),說明了棉絨的生長是由單基因座控制。研究中進而放大了QTL和GWAS的重疊區,發現了這個大約600 kb的區域包含推測的10個蛋白編碼基因(圖17 e)。在這一強烈的信號區域(-logP = 18.95)下或附近,發現了4個凱氏帶膜蛋白基因(casparian strip membrane protein genes)。在B-型細胞周期蛋白上游獲得了1個信號,而B-型細胞周期蛋白之前被報道過與毛狀體和纖維發育有關。

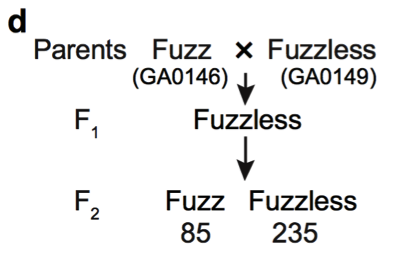
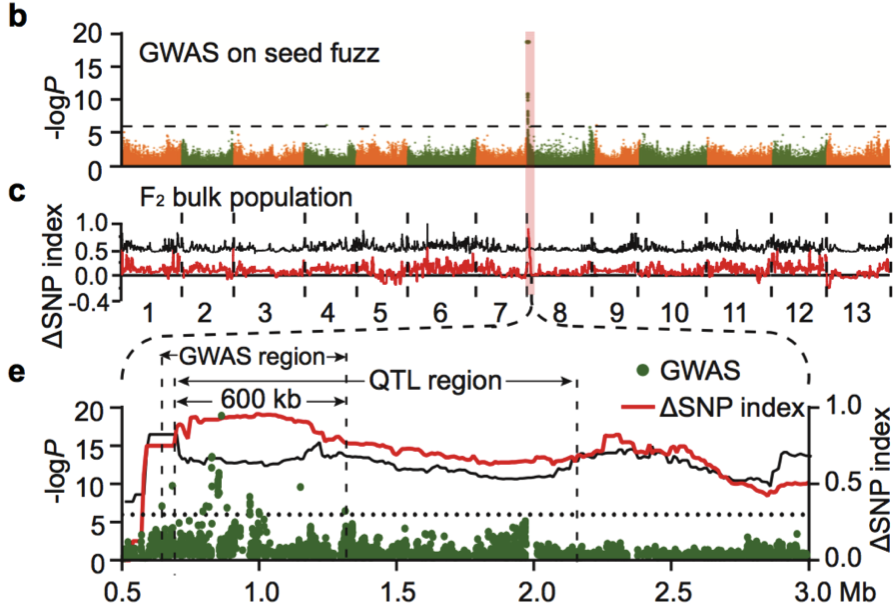
圖17 棉絨生長發育基因的聯合定位(GWAS+QTL)
a 亞洲棉種子有絨(左)和無絨(右)表型;b GWAS關聯分析;c F2群體QTL分析;d F2群體的構建 e GWAS和QTL聯合分析
亞洲棉在中國棉花的栽培史上具有重要的作用。本研究揭示了中國的亞洲棉群體呈現出不同的地理格局,這一觀點與其從中國的南部到長江和黃河的引入相一致。幾種表型如產量和抗病性狀在棉花從中國南部遷徙到長江再到黃河,經歷了顯著的變化,而這一變化受到了當地環境和人為選擇的影響。研究中通過不同種質群體的比較,獲得的地理受選擇的基因區域與QTL重疊區域是重要的,具高分辨率的遺傳資源,這將極大地促進棉花復雜性狀的改良。此外,研究中確定了GaKASIII基因可以促進棉花脂肪酸鏈的延伸和含油量,同時發現了2種典型的GaGSTF9基因單倍型啟動子與FOV抗性相關。最后結合GWAS與QTL共同定位的結果,鑒定了凱氏帶膜蛋白基因在棉絨細胞的發育過程中可能發揮功能性的作用。本研究表明地理隔離已經影響了SC,YZR和YER群體的遺傳基礎,同時影響了中國亞洲棉的抗病性和產量性狀的形成與分布。
想與我們的技術人員進行文章思路溝通?請點擊下面的按鈕,免費獲取設計方案。