直播內容安排
| 主題? | 直播內容? | 日期? | 會議鏈接? |
| 轉錄組班? | 有參轉錄組結題報告講解及常見數據挖掘? | 7.20(周二)? | 加入會議 |
| 轉錄組班? | 無參轉錄組結題報告講解及常見數據挖掘? | 7.27(周二)? | 加入會議 |
| 微生物班? | 微生物多樣性結題報告講解及常見數據挖掘? | 8.3(周二)? | 加入會議 |
| 微生物班? | 微生物多樣性APP操作及常見問題解答? | 8.10(周二)? | ?加入會議 |
更多課程上新中,敬請期待!!!
小福利~
每場直播中或直播后會抽取3名幸運聽眾好禮相送,以下獎品任選一項,即時生效:
- 轉錄組任意一款APP次賬號
- 微生物多樣性APP次賬號
- 百邁客1W云豆
每周二下午13:30與您相約百邁客云,助您數據挖掘!
本活動解釋權歸百邁客生物科技有限公司所有
]]>
新的一年,百邁客生物為了讓生物科研者更好的解讀基因、助力科研,在新春到來之際特舉行“恭賀新春、幸運抽獎”活動。
活動詳情如下:
即日起至2021年2月28日
一等獎:3000元項目代金券——100名
二等獎:1000元項目代金券——200名
三等獎:500元項目代金券——300名
四等獎:精美紀念禮品一份——400名
微信掃一掃參與抽獎

進入上方活動頁面,獲取個人專屬海報后,分享給5個好友參與抽獎活動,即可獲贈百邁客云課堂無門檻1000元培訓課程抵用券,免費在線學習300+培訓課程。
部分課程:

活動規則
1、每個用戶限1次抽獎機會。
2、代金券滿3萬可用,不與其他優惠活動同享,有效期至3月31日,中獎后代金券會自動存入微信“我—卡包—券和禮品卡”中,請您在有效期內聯系我們及時使用,所有代金券不得兌換現金,不設找零,僅可抵用一次。
3、紀念品將在活動結束后工作人員將與您取得聯系,溝通派送事宜。
本活動最終解釋權歸北京百邁客生物科技有限公司所有
]]>會員卡2:任選6個生信課程,7000元/年/人
會員卡3:任選10個生信課程,10000元/年/人
會員卡4:任選13個生信課程,12000元/年/人
會員權益
1、免費視頻包:百邁客云課堂現有價值近6千的收費課程免費看(除以上生信課程外)
2、免費直播課程:所選課程對應1年的直播觀看權限,觀看直播不限次。
3、免費線下活動:免費參加線下會員活動機會,會員活動每年至少一次,活動不僅有專家、有美食、有美酒同時還有美景與技術牛
4、會員卡3、4另贈送百邁客第八屆全國功能基因組學峰會門票(峰會為>500人會議,現已舉辦7屆,每屆均有院士大牛參加,收獲絕對多)
5、收費工具免費用:生信課程中同樣分析可選擇代碼或在線工具實現,其中收費在線工具可有3個月免費使用權
6、購課折扣(超過會員卡內的課程,每增加一個可享7折優惠)
7、所有會員卡需在購買后1月內選擇課程。
會員卡1、2所選的課程需在3個月內兌換開通;
會員卡3、4所選課程在購買后6個月內兌換開通。
注:1、所有課程有效期從開通之時開始計算。2、所選課程有未上線的,可在上線后2個月內選擇開通。若上線課程在兌換開通時間內,則此條無效。
8、課程視頻有效期一年
9、所有套餐僅限購買本人參加

注:表中直播時間為已確定的直播月份,后期會根據實際情況再次追加直播場次。
購買以上任意一個生信課程,均可享受課程一年視頻觀看及參加一次課程直播權限。購買百邁客試劑盒滿5000元,可贈送生信單課程一個

套餐選課推薦
1、生信入門基礎課:零基礎R語言繪圖+生信入門課程(可組合:轉錄組數據挖掘+代謝蛋白生信課程)
2、基因組類課程:比較基因組學+基因家族分析課程(可組合:零基礎R語言繪圖+生信入門+GWAS+遺傳進化+重測序分析課程)
3、群體類課程:GWAS+遺傳進化課程+重測序分析課程(可組合:零基礎R語言繪圖+生信入門+比較基因組+基因家族分析課程)
4、轉錄調控類課程:轉錄組數據挖掘+代謝蛋白組學+ATAC分析課程(可組合:零基礎R語言繪圖+生信入門)
5、微生物多樣性課程:微生物多樣性生信分析課程+微生物數據挖掘課程(可組合:零基礎R語言繪圖+生信入門)
具體課程介紹
一、零基礎R語言繪圖課程
本課程主要是用R代碼來介紹方法的實現,實操性強。同時,本次課程會系統性對R語言基礎進行講解,讓大家對R語言形成一個整體的思路。現在開源的R代碼很多,了解了基礎構建,后面拿到開源的代碼后就可以自己知道怎么修改,這些參數是什么了。對于后期R語言的自學是一個很好的敲門磚。

二、生信入門基礎課程
本課程主要針對沒有接觸過生信,而且沒有具體學習的生信方向,想了解入門生信的同學們。本課程涉及生信入門基礎的了解知識、常用的在線工具、以及涉及的幾種語言基礎介紹,讓你了解生信大概內容,從而根據這些內容了解自己想學習的方向,進而一步步細化去學習研究。

三、轉錄組數據挖掘生信課程
本課程主要針對轉錄組結果數據的挖掘分析,將從常用的轉錄組生信分析基礎入手,帶領大家學習轉錄組分析中常用的分析方案、個性化分析軟件及其實現方法,其中包含熱點的WGCNA分析,熱圖繪制,差異基因分類分析等繪圖分析。滿足轉錄組文章常見分析展示需求。

四、代謝與蛋白組學生信課程
本課程主要針對代謝和蛋白組學結果數據進行挖掘分析,從代謝和蛋白組學結果文件入手,手把手教授個性化繪圖及分析技巧。

五、比較基因組生信分析課程
本課程主要從基礎生信操作開始,從已發表的NG、NC等高水平文章入手,帶領大家學習并實際操作高分文章中圖形及分析是如何實現的,學習比較基因組學常用的分析方法,從而滿足文章后期發表的需求。

六、基因家族生信分析課程
近年來基因家族的生物信息學分析已經成為大家研究的熱點,目前已經表明許多重要的基因,如一些轉錄因子,都是以基因家族的形式存在。本課程系統的對基因家族進行了生物信息分析,讓大家更加深入學習了解基因家族分析研究,從而進一步豐富我們研究的內容,提升文章檔次,實現科研成果的快速產出。

七、GWAS生信分析課程
本課程主要從實踐入手,重點講解 GWAS 分析中用到的各種分析方法與常用分析軟件。包括群體結構分析、主成分分析、親緣關系分析、連鎖不平衡分析、單體型分析等內容。從原始數據開始,逐步完成分析內容,并對分析內容進行整理與挖掘,將數據轉化成更直觀的圖形來展示最終的分析結果。若您想做、準備做或正在做GWAS相關的項目,那這個課程就很適合您,即可學會自己分析,同時了解分析過程中的參數設置,可以很好協助您的論文寫作。

八、微生物多樣性生信分析課程
本課程將微生物多樣性生信分析中的每一個步驟以及每一步驟所涉及的參數都很明確的列了出來,詳細為大家進行講解及實操演示,幫助大家更好的了解學習微生物多樣性分析的整個過程。

九、微生物多樣性數據挖掘分析課程
本次課程主要針對微生物多樣性數據的后期挖掘繪圖,主要利用R語言來進行微生物多樣性相關圖形的繪制,從而實現自己可以分析繪制圖形,更重要的是自己可以根據不同雜志的要求,很快得出對應的圖形。

十、重測序生信分析課程
本課程主要針對基于二代及三代測序的重測序分析。以實際操作為主,從序列比對開始到突變位點的分析及應用。包括常用的分析方法及工具軟件(BWA、GATK、Plink等)。讓大家能夠快速上手有關重測序的相關分析并為后續與突變相關的高級分析打下良好的基礎。

十一、遺傳進化生信分析課程
本次課程主要講解群體遺傳進化分析中常見分析內容以及定制化繪制相關圖形,同時涵蓋高級分析,基因流、分化時間、群體歷史動態三塊高級分析內容。本課程主要面向初中級,且對遺傳進化分析有需求的老師。

十二、ATAC生信分析課程
本課程主要從技術原理、實驗步驟、生物信息學分析、文獻分享四大內容向大家介紹ATAC-Seq技術。同時,本課程還涉及專門的生物信息學實操課程,帶領大家學習ATAC-Seq基本分析流程,其中包括數據質控、Peak Calling、Motif分析等。從而幫助大家將ATAC-Seq運用在自己的課題當中。

基于樣品中的物種組成及豐度信息推測樣品中表型類型和功能組成及差異。包含PICRUSt2功能預測、FAPROTAX功能預測、BugBase表型預測、Tax4Fun2功能預測和真菌FunGuild表型預測。
一. PICRUSt2功能預測
PICRUSt1軟件依賴于Greengene數據庫進行物種比對、功能數據輸出。而Greengene數據庫在2013年之后就停止了更新,距今為止已有7年。隨著時間的推移,大量微生物基因組數據測序獲得,而停止更新的Greengene數據庫限制了PICRUSt的功能預測范圍。對于近年來測序獲得微生物功能功能信息無法進行預測,滿足不了當前的研究需求,為了補上這塊滿足科研工作者的需求,PICRUSt團隊于近期升級了軟件,正式公布了PICRUSt2。
1)將待預測的OTU代表序列置于軟件中已有的系統發育樹中,而不是直接對OTU序列進行分類學注釋;
2)不再基于GreenGene 16S數據庫進行功能預測,其用于預測的參考基因組數據庫相比先前也已擴大了10倍以上

參考文獻:Douglas G M, Maffei V J, Zaneveld J, et al. PICRUSt2: An improved and extensible approach for metagenome inference. bioRxiv, 2019.
功能組成分析:統計各樣品在不同分類層級上的功能組成。

注:橫坐標為樣品名稱;縱坐標為功能相對豐度百分比。
功能差異分析:統計各樣品或者各組在不同分類層級上的功能差異。

注:圖中不同顏色代表不同的樣品或分組。左圖所示為不同功能在兩個樣品或者兩組樣品中的豐度比例,中間所示為95%置信度區間內功能豐度的差異比例,最右邊的值為校正后p值。
二. FAPROTAX功能預測
FAPROTAX較適用于對環境樣本的生物地球化學循環過程(特別是碳、氫、氮、磷、硫等元素循環)進行功能注釋預測。FAPROTAX是根據已發表的文獻手動構建的數據庫,它把原核微生物的分類和代謝等功能對應起來,目前收集自4600多個原核微生物的80多個功能分組7600多條功能注釋信息。

參考文獻:Louca S, Parfrey L W, Doebeli M. Decoupling function and taxonomy in the global ocean microbiome[J]. Science, 2016, 353(6305): 1272-1277.
三. BugBase表型預測
BugBase是一種預測復雜微生物組內功能途徑的生物水平覆蓋以及生物可解釋表型的方法。BugBase首先通過預測的16S拷貝數對OTU進行歸一化,然后使用提供的預先計算的文件預測微生物表型。包括以下七方面:革蘭氏陽性 (Gram Positive)、革蘭氏陰性 (Gram Negative)、生物膜形成 (Biofilm Forming)、致病潛力 (Pathogenic Potential)、移動元件含量 (Mobile Element Containing)、氧的利用 (Oxygen Utilizing)、氧化脅迫耐受 (Oxidative Stress Tolerant)。
參考文獻:Ward T, Larson J, Meulemans J, Hillmann B, Lynch J, SidiropoulosD,Spear J, Caporaso G, Blekhman R, Knight R, Fink R, Knights D. 2017.BugBase predicts organism level microbiome phenotypes. bioRxiv.
四. Tax4Fun2功能預測
Tax4Fun全面升級為Tax4Fun2.評估微生物群落的功能和冗余度是環境微生物學的主要挑戰。Tax4Fun2可基于16S rRNA基因序列快速預測原核生物的功能譜和功能冗余。通過合并用戶定義的、特定于棲息地的基因組信息,可以顯著提高預測功能圖譜的準確性。
優點:
(1)不再局限于僅SILVA的特定版本注釋的OTU豐度表,允許直接以OTU代表序列作為輸入,通過與指定參考數據庫的比對實現物種注釋。除了Tax4Fun2提供的已構建好的參考集(相比之前大幅擴大),也允許我們提供自定義的參考集,使用非常靈活。
(2)側重于原核數據,但也可以合并真核數據。
(3)提供了計算特定功能冗余的方法,對于預測特定功能在環境擾動期間丟失的可能性至關重要。
(4)精度和穩定性顯著提升。

參考文獻:Wemheuer F, Taylor J A, Daniel R, et al. Tax4Fun2: a R-based tool for the rapid prediction of habitat-specific functional profiles and functional redundancy based on 16S rRNA gene marker gene sequences. bioRxiv, 2018.
五. FunGuild表型預測
FUNGuild(Fungi Functional Guild)是一種可用于由生態協會分類學解析真菌的工具,用簡單而一致的方法將大型序列庫分類為具有生態意義的類別。根據營養方式將真菌分為12類,然后構建了一個真菌分類和功能分組(guild)之間的數據庫,通過這個數據庫你就可以對真菌進行功能分類。
病理營養型(pathotroph):通過損害宿主細胞而獲取營養(包括吞噬型真菌phagotrophs)。
共生營養型(symbiotroph):通過與宿主細胞交換資源來獲取營養。
腐生營養型(saprotroph):通過降解死亡的宿主細胞來獲取營養。包括動物病原菌(animal pathogens)、叢枝菌根真菌(arbuscular mycorrhizal fungi)、外生菌根真菌(ectomycorrhizal fungi)、杜鵑花類菌根真菌(ericoid mycorrhizal fungi)、葉內生真菌(foliar endophytes)、地衣寄生真菌(lichenicolous fungi)、地衣共生真菌(lichenized fungi)、菌寄生真菌(mycoparasites)、植物病原菌(plantpathogens)、未定義根內生真菌(undefined root endophytes)、未定義腐生真菌(undefined saprotrophs)和木質腐生真菌(wood saprotrophs)。
參考文獻:Nguyen NH, Song Z, Bates ST, Branco, S, Tedersoo L, Menke J, Schilling JS, Kennedy PG. 2016. FUNGuild: an open annotation tool for parsing fungal community datasets by ecological guild. Fungal Ecology 20:241-248.
MicroPITA分析
在做完16S、18S或ITS等微生物多樣性研究后,我們常常還會想進一步了解微生物群落的功能。通常情況下,會采用宏基因組、宏轉錄組或宏代謝組等方法深入分析,但相對于擴增子測序,宏基因組等測序手段的價格還是相對較高,因此需要從已測完的樣本中再挑選合適的樣本進行宏基因組測序。可利用microPITA進行樣品預測,挑選出合適的樣品。該分析是基于大量微生物多樣性的數據,根據不同指標篩選出代表性樣本,以便于開展開展后續研究。
| 類型 | 方法 | 含義 | 樣本特點 |
| 無監督方法 | diverse | 選擇α多樣性最高的樣本 | 生態多樣性高 |
| features | 根據目標物種挑選樣本 | 針對特定物種 | |
| extreme | 選擇β多樣性距離最遠的樣本 | 極端樣本 | |
| representative | 最能反映整體距離差異的樣本 | 核心樣本 | |
| 有監督方法 | Distinct | 根據表型/分組特征,挑選組間β多樣性距離最大的樣本 | 依據表型/分組特征,選擇極端樣本 |
| Discriminant | 根據表型/分組特征,挑選離分組中心最近的樣本 | 依據表型/分組特征,挑選核心樣本 |
參考文獻:Tickle TL, Segata N, Waldron L, Weingart U, Huttenhower C. Two-stage microbial community experimental design. ISME J. 2013 Dec;7(12):2330-9. doi: 10.1038/ismej.2013.139. Epub 2013 Aug 15. PMID: 23949665; PMCID: PMC3834858.
群落結構分析
微生物群落生態學的一個主要目標是了解構成跨時空物種豐度模式的過程。確定性和隨機性兩種類型的過程會影響群落的聚集。確定性過程與生態選擇相關,隨機過程包括不可預測的擾動、概率性的散布和隨機的出生-死亡事件等,這些變化不是由環境決定的適應性結果。通過零模型量化群落的絕對系統發育距離與隨機系統發育距離的偏離度,偏離程度越大,群落受確定性因素的影響越大,偏離度越小,群落受隨機性因素的影響越大。通常使用βNTI(最近種間親緣關系指數)以評估不同時空尺度下隨機性和確定性過程對微生物群落組裝的影響。

其中,| βNTI |>2表示觀察到的兩個群落之間的更替主要由選擇控制,其中βNTI>+2與變量選擇一致,而βNTI<-2表示同質選擇。因此,| βNTI |<2意味著一組群落的更替受擴散限制、均勻化擴散或未消除過程的控制。為了理清這些過程,Raup-Crick矩陣(RCbray)基于群落的標準Bray-Curtis矩陣構建,提供有關所觀察到的流動程度是否明顯偏離預期的信息。這個值等于觀測到的Bray-Curtis和零分布之間的偏差,范圍是-1到+1。| RCbray |<0.95可以解釋為終止過程的影響。反過來,擴散限制加上漂移導致大于預期的周轉率(RCbray>+0.95),而RCbray<-0.95則表明群落組成的周轉率主要受均勻擴散控制。
參考文獻:Jizhong, Zhou, Daliang, et al. Stochastic Community Assembly: Does It Matter in Microbial Ecology[J]. Microbiology & Molecular Biology Reviews, 2017.
]]>
發表期刊:Plant Biotechnology Journal
發表時間:2020年7月12
影響因子:8.1
研究內容:棉花花芽分化
研究對象:兩個早熟品種,兩個晚熟品種;每個品種6個時期
研究方法:轉錄組測序
前言
陸地棉(Gossypium hirsutum L.)是世界上最重要的纖維作物。在我國黃河流域和長江流域棉區,實現早熟棉在小麥或者油菜后直播可以提高復種指數。而在西北內陸棉區,春季氣溫低,秋季枯霜早,早熟棉既能提高棉花的霜前花率,還可以改善棉花品質。花芽分化是影響短季棉品種早熟的重要性狀,是棉花現蕾期、開花期和成鈴期發育的基礎,花芽分化直接影響開花時間。花芽分化是植物從營養生長向生殖生長過渡的標志。當進入生殖生長時,側芽變成花芽,花芽發育成果枝。果枝分化決定了開花量、生殖能力和棉花產量。因此,早熟性狀及早熟棉品種在生產上顯得尤為重要。
結果
1、棉花莖尖的形態發育

圖1兩個早熟品種和兩個晚熟品種的比較
2、棉花不同發育階段的72個RNA文庫的轉錄組譜

?圖2早熟和晚熟品種六個發育階段之間的轉錄關系。
3、加權基因共表達網絡分析(WGCNA)

?圖3差異表達基因的WGCNA
4、棉花從營養生長向生殖生長轉換相關基因的鑒定

?圖4模塊MElightcyan的分析
5、GhCAL在調節從營養生長到生殖生長的轉換的功能驗證





百邁客云平臺可以在所有基因挖掘這里進行所有基因篩選和PCA聚類(動圖是示例數據)
然后作者利用高分文章利器:WGCNA分析,進行數據深度挖掘和整理,推薦大家使用百邁客云平臺小工具WGCNA模塊,可以出來和作者同樣高大上的圖片。
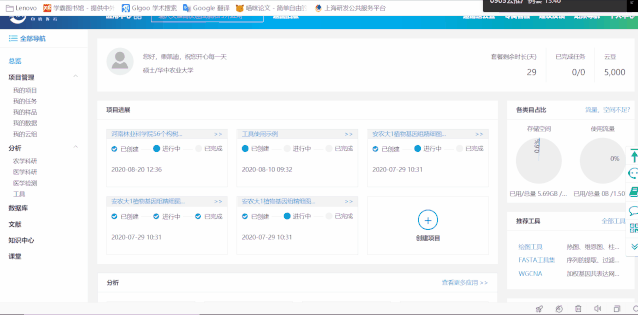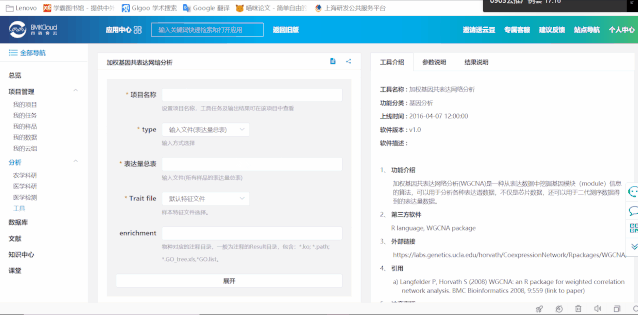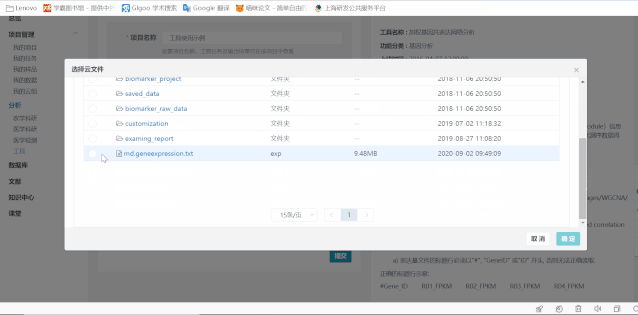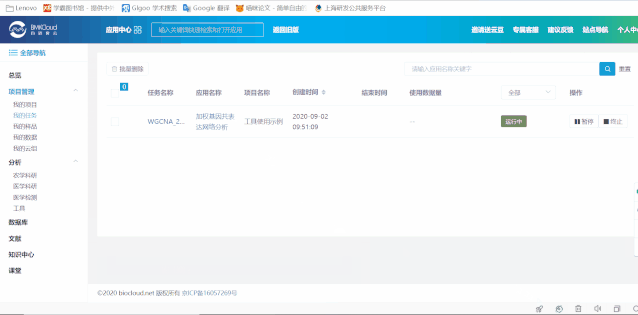
登陸百邁客云平臺—小工具,即可使用分析(動圖是示例數據)
然后作者根據一系列的功能驗證最終鎖定控制花芽分化的基因,再次推薦大家使用百邁客轉錄組個性化(所有基因挖掘,差異基因挖掘,基因結構挖掘)三大模塊和小工具(108款分析繪圖工具),下一篇高分文章就是你!

作為一個科技服務工作者,自然能夠明白每一位老師的痛處和難點,不過現在都2020年了,再也不是一個轉錄組1萬元的天價了,那么現在從原始的測序數據到數據挖掘直至最后完美的SCI論文圖表,究竟是怎么出來呢?
雖然君子遠庖廚,不過今天小編將為您帶進后廚,為您娓娓道來。
1、首先我們需要原始數據
2、一分鐘的分析任務投遞
選擇合適的分析APP

命名和選擇數據


選擇參考基因組及設置差異分組


任務提交后,根據樣本數據量,一般24-48h左右大家就可以看到一份完整的分析結題報告和分析數據了。
講到這里可能有人會問:難道就這么簡單?那人家好幾篇文章里面那些高大上的圖片都是大神用小工具做的嗎,小編可以負責任的告訴你們,不是的!我們還有很多隱藏功能:
第一:基因檢索。(小編選的蛋白是PPR蛋白,從4萬多基因里面篩選出來60個PPR蛋白相關的基因,然后根據60個基因做GO分類圖)。

第二:WGCNA分析。這個分析主要是將基因模塊與表型數據或者表型樣本進行關聯,從而快速的鎖定一批候選基因。

運行后打開是下圖這樣的,對此圖有疑惑的可以點擊圖片左側的攝像頭。
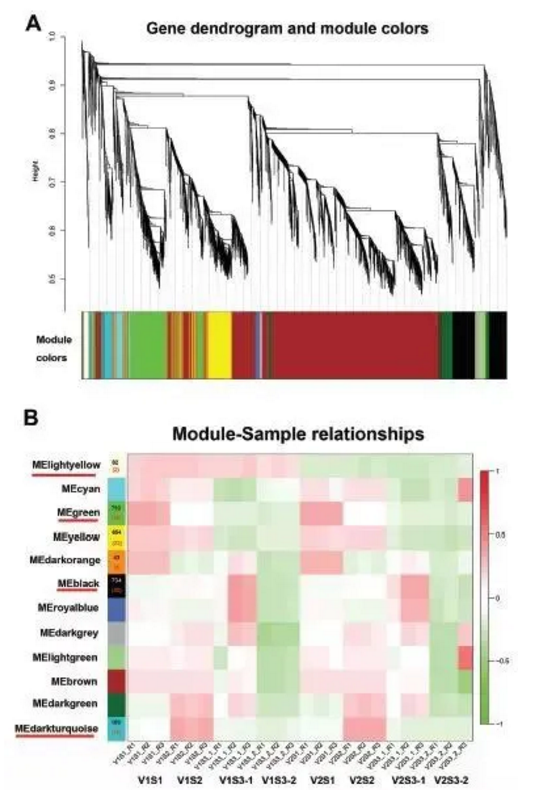
第三:最近很火的差異基因表達趨勢分析


第四:108款分析繪圖工具(73款常用工具免費使用)。

具體這些工具如何使用?百邁客云還有哪些隱藏功能呢?歡迎大家持續關注,小編會定期為大家進行分享。
]]>
下載谷歌瀏覽器,輸入網址https://international.biocloud.net/zh/user/login?,進入百邁客云平臺登錄界面,輸入賬號密碼登錄。賬號為手機號或者是郵箱,初始密碼為123.bmk.

二、選擇合適的分析平臺
百邁客云包含農學和醫學兩大類分析平臺,醫學分析平臺主要針對人、鼠數據的分析,包含:轉錄組、非編碼RNA、單基因病外顯子、腫瘤外顯子、重測序等數據分析,以及全轉錄組聯合分析;農學分析平臺可以應用到更多的物種,涵蓋轉錄組、非編碼RNA、微生物、蛋白、代謝等數據的分析,以及全轉錄組聯合分析等。點擊左側導航分析->農學或者醫學打開分析平臺列表頁面,點擊選擇您想要使用的分析平臺,打開其詳細介紹頁面,該頁面可以看到該平臺的應用領域“平臺介紹“技術背景“案例“課堂“版本記錄,點擊打開軟件即可進入到參數頁面。


三、創建項目名稱
為了方便數據、任務和報告的管理,我們將同屬于一個項目的內容會放到一個項目中,因此進行基本分析時需要先選擇一個項目,如果沒有項目也可以點擊+新建先創建一個項目,見下圖。

四、數據導入
針對FASTQ測序數據,選擇文件夾批量導入,雙端測序數據必須分別以_1.fq和_2.fq結尾,系統便會自動對數據進行配對,而且多個目錄中的文件可以分多次導入進來一起進行分析。(在本公司進行測序的數據會自動推送到您的賬戶下,數據所在文件夾名稱為合同編號)
導入數據之后,可以根據自己的需要修改樣品ID,由于此處設置的ID會體現在分析報告和分析結果中,因此請慎重考慮后再設置,分析完成后不可再次修改。如果平臺上還沒有您自己的數據,請參考數據上傳先將您的數據傳到云平臺上。


四、基本參數設置
一般在這里選擇生信流程版本、設置報告名稱等基本參數,如果對于參數設置沒有特殊要求,推薦使用默認參數。

五、選擇參考基因組(有參)
對于依賴參考基因組序列進行分析的平臺,一定要選擇和分析數據對應的參考物種及組裝版本,不同版本的參考基因組的詳細信息可以點擊基因組版本詳情進行查看。(一個項目只能使用一個版本的參考基因組進行分析)

六、基因功能注釋(無參)
- 組裝方式:
轉錄組組裝方式決定了后續Unigenes庫的構建和表達定量的策略以及分析結果的可靠性.根據實際情況選擇轉錄組組裝方式。分開組裝是對每個樣品數據單 獨組裝;合并組裝是將所有樣品放在一起組裝;分組組裝適合于不同品種(或者是變異種)的組裝,將相同品種的樣品合并組裝,然后將每組的組裝結果 進行合并去冗余。合并組裝獲得的Unigene庫更完整、冗余度更低,因此Trinity官方亦推薦使用合并組裝,以便進行后續的表達定量和差異表達分析.
- 注釋物種
為了提高注釋分析的效率(縮短比對比對時間)以及獲得有效的注釋信息,在選擇注釋物種時,應盡量選擇包含物種最確切的數據庫.(如果分析物種為真菌類,一定要選擇真菌選項,否則會影響組裝效果)

七、差異分組設置
根據實驗方案設計以及樣品信息,進行分組的設置,流程據此進行差異比較分析,此處只支持兩組間比較,可添加多個差異分組。FDR值一般推薦選擇0.01,差異倍數閾值一般推薦選擇(此處的參數和差異分組在后期報告個性化中可再次進行修改)

八、生成標準分析報告
參數設置完成之后,可以點擊保存參數,方便之后進行重新分析或者基于本次參數進行修改后再次分析;一切準備就緒后,點擊提交將任務提交到百邁客云計算集群上,根據不同分析平臺、不同數據量等待大概2小時到3周的時間完成項目分析,獲取到標準分析報告。
報告查看,點擊項目(管理)?->?我的項目打開項目列表,找到之前提交任務時選擇的項目,點擊項目名稱打開該項目,即可看到新生成的分析報告記錄,點擊報告名稱查看詳細報告。
點擊報告右上角的項目結果下載,可以選擇下載HTML報告、PDF報告和結果數據(尾款結清后)。HTML報告只包括分析報告的html文件及一個src文件夾,展示了部分結果;PDF報告是根據HTML報告轉換而來,方便您進行報告打印;結果數據包含了項目中所有結果文件,一般比較大,會通過FTP進行下載,請耐心等待下載。


下載谷歌瀏覽器,輸入網址https://international.biocloud.net/zh/user/login?,進入百邁客云平臺登錄界面,輸入賬號密碼登錄。賬號為手機號或者是郵箱,初始密碼為123.bmk.
二、選擇合適的分析平臺
百邁客云包含農學和醫學兩大類分析平臺,醫學分析平臺主要針對人、鼠數據的分析,包含:轉錄組、非編碼RNA、單基因病外顯子、腫瘤外顯子、重測序等數據分析,以及全轉錄組聯合分析;農學分析平臺可以應用到更多的物種,涵蓋轉錄組、非編碼RNA、微生物、蛋白、代謝等數據的分析,以及全轉錄組聯合分析等。點擊左側導航分析->農學或者醫學打開分析平臺列表頁面,點擊選擇您想要使用的分析平臺,打開其詳細介紹頁面,該頁面可以看到該平臺的應用領域“平臺介紹“技術背景“案例“課堂“版本記錄,點擊打開軟件即可進入到參數頁面。

三、創建項目名稱
為了方便數據、任務和報告的管理,我們將同屬于一個項目的內容會放到一個項目中,因此進行基本分析時需要先選擇一個項目,如果沒有項目也可以點擊+新建先創建一個項目,見下圖。

四、數據導入
針對FASTQ測序數據,選擇文件夾批量導入,雙端測序數據必須分別以_1.fq和_2.fq結尾,系統便會自動對數據進行配對,而且多個目錄中的文件可以分多次導入進來一起進行分析。(在本公司進行測序的數據會自動推送到您的賬戶下,數據所在文件夾名稱為合同編號)
導入數據之后,可以根據自己的需要修改樣品ID,由于此處設置的ID會體現在分析報告和分析結果中,因此請慎重考慮后再設置,分析完成后不可再次修改。如果平臺上還沒有您自己的數據,請參考數據上傳先將您的數據傳到云平臺上。
!注:1.各組學導入數據樣本個數要一致
????2.circRNA數據選擇去核糖體建庫則與lncRNA共用一套數據,無需單獨導入(百邁客建庫方法為去核糖體建庫);選擇去線性建庫則需要單獨導入數據


五、選擇樣本對應關系
為了方便后面的聯合分析內容,需要將各RNA項目里的數據按照對應關系統一編號,有幾組對應關系則在左側添加幾組,再從右側的lncRNA樣品池和miRNA樣品池中選擇對應的樣本添加到分組。其中默認按鈕可以按照數字的順序快速添加對應關系。

六、選擇參考基因組
對于依賴參考基因組序列進行分析的平臺,一定要選擇和分析數據對應的參考物種及組裝版本,不同版本的參考基因組的詳細信息可以點擊基因組版本詳情進行查看。如果云上沒有您需要的參考基因組,您可以聯系對應的運營將您提供的基因組文件部署到云平臺上,供您使用。(注:一個項目只能使用一個版本的參考基因組進行分析)

七、參數設置
- 流程版本號可以選擇默認的最新版本
Lib_type為窗體頂端
- Lib_type :lncRNA建庫方式,fr-firststrand表示測序數據中,reads2方向與轉錄本方向一致;fr-unstranded為非鏈特異性建庫;fr-secondstrand 表示read1方向與轉錄本方向一致(百邁客lncRNA的建庫方式為fr-firststrand)
- lncRNA分析中反式靶基因預測方法: 樣本少的時候可以選擇基于序列方法,樣本數較多(大于5)推薦使用基于共表達分析方法。
- miRNA的長度范圍和接頭序列可根據實際情況進行填寫,主要是為了過濾原始數據中的街頭,默認參數為百邁客使用的序列
- circRNA預測軟件:給出三種預測方法,CIRI和find_circ是兩個不同的預測軟件,可以分別選擇其中一個軟件進行預測,CIRI+find_circ是將兩個軟件預測的結果取交集作為最終的結果,(此選項可能會導致預測結果偏少)

八、差異分組設置
根據實驗方案設計以及樣品信息,進行分組的設置,流程據此進行差異比較分析,此處只支持兩組間比較,可添加多個差異分組。
DEseq2軟件適用于有生物學重復的項目,edgeR適用于無生物學重復的項目,選擇第一個,系統會自動識別項目類型選擇對應的軟件
- FDR值一般推薦選擇0.01,差異倍數閾值一般推薦選擇2.(此處設置的的參數和差異分組在后期報告個性化中可再次進行修改)

九、聯合分析參數
- 構建共表達網絡的篩選條件為共表達相關性閾值和共表達相關性分析中顯著性閾值兩個參數,其中共表達相關性閾值越大,顯著性閾值越小,則條件越嚴格。
- 構建ceRNA網絡的關系對的篩選條件為ceRNA超幾何檢驗fdr值、ceRNA超幾何檢驗p值、ceRNA共享的miRNA數目三個參數,其中fdr值和P值越小,共享miRNA數目越大則篩選條件越嚴格。
- 圖中參數為推薦參數

十、生成標準分析報告
參數設置完成之后,可以點擊保存參數,方便之后進行重新分析或者基于本次參數進行修改后再次分析;一切準備就緒后,點擊提交將任務提交到百邁客云計算集群上,根據不同分析平臺、不同數據量等待大概2小時到3周的時間完成項目分析,獲取到標準分析報告。
報告查看,點擊項目(管理)?->?我的項目打開項目列表,找到之前提交任務時選擇的項目,點擊項目名稱打開該項目,即可看到新生成的分析報告記錄,點擊報告名稱查看詳細報告。
點擊報告右上角的項目結果下載,可以選擇下載HTML報告、PDF報告和結果數據(尾款結清后)。HTML報告只包括分析報告的html文件及一個src文件夾,展示了部分結果;PDF報告是根據HTML報告轉換而來,方便您進行報告打印;結果數據包含了項目中所有結果文件,一般比較大,會通過FTP進行下載



下載谷歌瀏覽器,輸入網址https://international.biocloud.net/zh/user/login?,進入百邁客云平臺登錄界面,輸入賬號密碼登錄。賬號為手機號或者是郵箱,初始密碼為123.bmk.
二、選擇合適的分析平臺
百邁客云包含農學和醫學兩大類分析平臺,醫學分析平臺主要針對人、鼠數據的分析,包含:轉錄組、非編碼RNA、單基因病外顯子、腫瘤外顯子、重測序等數據分析,以及全轉錄組聯合分析;農學分析平臺可以應用到更多的物種,涵蓋轉錄組、非編碼RNA、微生物、蛋白、代謝等數據的分析,以及全轉錄組聯合分析等。點擊左側導航分析->農學或者醫學打開分析平臺列表頁面,點擊選擇您想要使用的分析平臺,打開其詳細介紹頁面,該頁面可以看到該平臺的應用領域“平臺介紹“技術背景“案例“課堂“版本記錄,點擊打開軟件即可進入到參數頁面。

三、創建項目名稱
為了方便數據、任務和報告的管理,我們將同屬于一個項目的內容會放到一個項目中,因此進行基本分析時需要先選擇一個項目,如果沒有項目也可以點擊+新建先創建一個項目,見下圖。

四、選擇輸入文件
- 蛋白鑒定表格:蛋白質質譜鑒定的結果表格。制表符分隔的文本文件,第一列為蛋白ID,其他列為樣品的表達量。對格式不清楚的可點擊查看示例下載示例文件。(百邁客項目的結果文件會直接推送到客戶賬號下,文件夾名稱為合同編號,可直接導入使用)
- 蛋白數據庫:蛋白質質譜鑒定所用的數據庫。不同數據庫得到的蛋白ID格式不同。這里給出了常用的uniport數據庫和轉錄組數據庫。如果需要和轉錄組聯合分析,建議使用轉錄組數據庫,并且可以輸入轉錄組的注釋信息文件,百邁客的轉錄組項目會自動生成此文件,也可以下載示例查看具體的格式)。

五、綜合選項
- 流程版本:選擇最新的版本(目前為0)
- Kegg注釋物種分類:點擊下拉框選擇KEGG注釋所用的物種類別
- 報告名稱:可以自定義方便區分的報告名稱,可以使用默認名稱
- 物種名稱:支持自定義物種名稱,與后期生成的任務名稱相關聯
- 標準化方式:否:不進行標準化;總峰面積:表示每個樣本中的每個蛋白除以該樣本總的峰面積;加和方法:先計算單個樣本所有蛋白的sum值, 再用sum值除以幾個樣本sum值中最大的,得到偏差系數,最后用蛋白/樣本的偏差系數得到標準化后的值。此處iTRAQ項目數據已經進行過歸一化,可以選擇否,lable-free項目推薦總峰面積

六、差異分組設置
根據實驗方案設計以及樣品信息,進行分組的設置,流程據此進行差異比較分析,此處只支持兩組間比較,可添加多個差異分組。
- pvalue值一般推薦選擇0.01,差異倍數閾值一般推薦選擇2.(此處設置的的參數和差異分組在后期報告個性化中可再次進行修改)
- 分組設置:有幾組生物學重復就在左側的分組池中添加相應數量的分組,再從右側的樣品池中選樣本加入相應的分組中,1個樣本只能屬于一個分組且一個分組的樣本數目不少于2個(這里需要修改分組名稱,分析完成后不可修改)
- 差異分組設置
根據實驗方案設計以及樣品信息,進行分組的設置,流程據此進行差異比較分析,此處只支持兩組間比較,可添加多個差異分組。(此處的參數和差異分組在后期報告個性化中可再次進行修改)

七、生成標準分析報告
參數設置完成之后,可以點擊保存參數,方便之后進行重新分析或者基于本次參數進行修改后再次分析;一切準備就緒后,點擊提交將任務提交到百邁客云計算集群上,根據不同分析平臺、不同數據量等待大概2小時到3周的時間完成項目分析,獲取到標準分析報告。
報告查看,點擊項目(管理)?->?我的項目打開項目列表,找到之前提交任務時選擇的項目,點擊項目名稱打開該項目,即可看到新生成的分析報告記錄,點擊報告名稱查看詳細報告。
點擊報告右上角的項目結果下載,可以選擇下載HTML報告、PDF報告和結果數據(尾款結清后)。HTML報告只包括分析報告的html文件及一個src文件夾,展示了部分結果;PDF報告是根據HTML報告轉換而來,方便您進行報告打印;結果數據包含了項目中所有結果文件,一般比較大,會通過FTP進行下載,請耐心等待下載。
]]>

下載谷歌瀏覽器,輸入網址https://international.biocloud.net/zh/user/login?,進入百邁客云平臺登錄界面,輸入賬號密碼登錄。賬號為手機號或者是郵箱,初始密碼為123.bmk.
二、選擇合適的分析平臺
百邁客云包含農學和醫學兩大類分析平臺,醫學分析平臺主要針對人、鼠數據的分析,包含:轉錄組、非編碼RNA、單基因病外顯子、腫瘤外顯子、重測序等數據分析,以及全轉錄組聯合分析;農學分析平臺可以應用到更多的物種,涵蓋轉錄組、非編碼RNA、微生物、蛋白、代謝等數據的分析,以及全轉錄組聯合分析等。點擊左側導航分析->農學或者醫學打開分析平臺列表頁面,點擊選擇您想要使用的分析平臺,打開其詳細介紹頁面,該頁面可以看到該平臺的應用領域“平臺介紹“技術背景“案例“課堂“版本記錄,點擊打開軟件即可進入到參數頁面。

三、創建項目名稱
為了方便數據、任務和報告的管理,我們將同屬于一個項目的內容會放到一個項目中,因此進行基本分析時需要先選擇一個項目,如果沒有項目也可以點擊+新建先創建一個項目,見下圖。

四、選擇輸入文件
代謝定量文件:針對鑒定到的代謝物進行定量的結果表格。制表符分隔的文本文件,第一列為代謝物ID,其他列為樣品的表達量。對格式不清楚的可點擊查看示例下載示例文件。(百邁客項目的結果文件會直接推送到客戶賬號下,文件夾名稱為合同編號,可直接導入使用)

五、綜合選項
- 流程版本:選擇最新的版本(目前為0)
- Kegg注釋物種分類:點擊下拉框選擇KEGG注釋所用的物種類別
- 報告名稱:可以自定義方便區分的報告名稱,可以使用默認名稱
- 物種名稱:支持自定義物種名稱,與后期生成的任務名稱相關聯
- 標準化方式:否表示不使用歸一化,總峰面積歸一化表示每個樣本中的每個代謝物除以該樣本總的峰面積,內標則是每個代謝物除以內標代謝物的峰面積且默認內標的代謝物定性名稱為IS,內標不參與后續的分析。(LC推薦使用總峰面積,GC推薦使用內標)

六、差異分組設置
根據實驗方案設計以及樣品信息,進行分組的設置,流程據此進行差異比較分析,此處只支持兩組間比較,可添加多個差異分組。
- 推薦閾值選擇:(1)差異倍數閾值1、T檢驗p值0.05、VIP值1 (2)差異倍數閾值2、T檢驗p值1、VIP值1
(其中vip值采用了正交偏最小二乘法判別分析(OPLS-DA),該分析會給每個代謝物一個變量投影重要度VIP值,值越大說明代謝物的差異越顯著)
- 分組設置:有幾組生物學重復就在左側的分組池中添加相應數量的分組,再從右側的樣品池中選樣本加入相應的分組中,1個樣本只能屬于一個分組且一個分組的樣本數目不少于2個。(這里需要修改分組名稱,分析完成后不可修改)
- 差異分組設置:根據實驗方案設計以及樣品信息,進行分組的設置,流程據此進行差異比較分析,此處只支持兩組間比較,可添加多個差異分組。(此處的參數和差異分組在后期報告個性化中可再次進行修改)

七、生成標準分析報告
參數設置完成之后,可以點擊保存參數,方便之后進行重新分析或者基于本次參數進行修改后再次分析;一切準備就緒后,點擊提交將任務提交到百邁客云計算集群上,根據不同分析平臺、不同數據量等待大概2小時到3周的時間完成項目分析,獲取到標準分析報告。
報告查看,點擊項目(管理)?->?我的項目打開項目列表,找到之前提交任務時選擇的項目,點擊項目名稱打開該項目,即可看到新生成的分析報告記錄,點擊報告名稱查看詳細報告。
點擊報告右上角的項目結果下載,可以選擇下載HTML報告、PDF報告和結果數據(尾款結清后)。HTML報告只包括分析報告的html文件及一個src文件夾,展示了部分結果;PDF報告是根據HTML報告轉換而來,方便您進行報告打印;結果數據包含了項目中所有結果文件,一般比較大,會通過FTP進行下載,請耐心等待下載。
]]>